CLI Pipeline Walkthrough¶
This page is the user’s guide to the end-to-end Cherimoya CLI: how to take raw BAM/BED files and a reference genome through peak calling, training, attribution, seqlet calling, motif discovery, and motif marginalization in a single reproducible run.
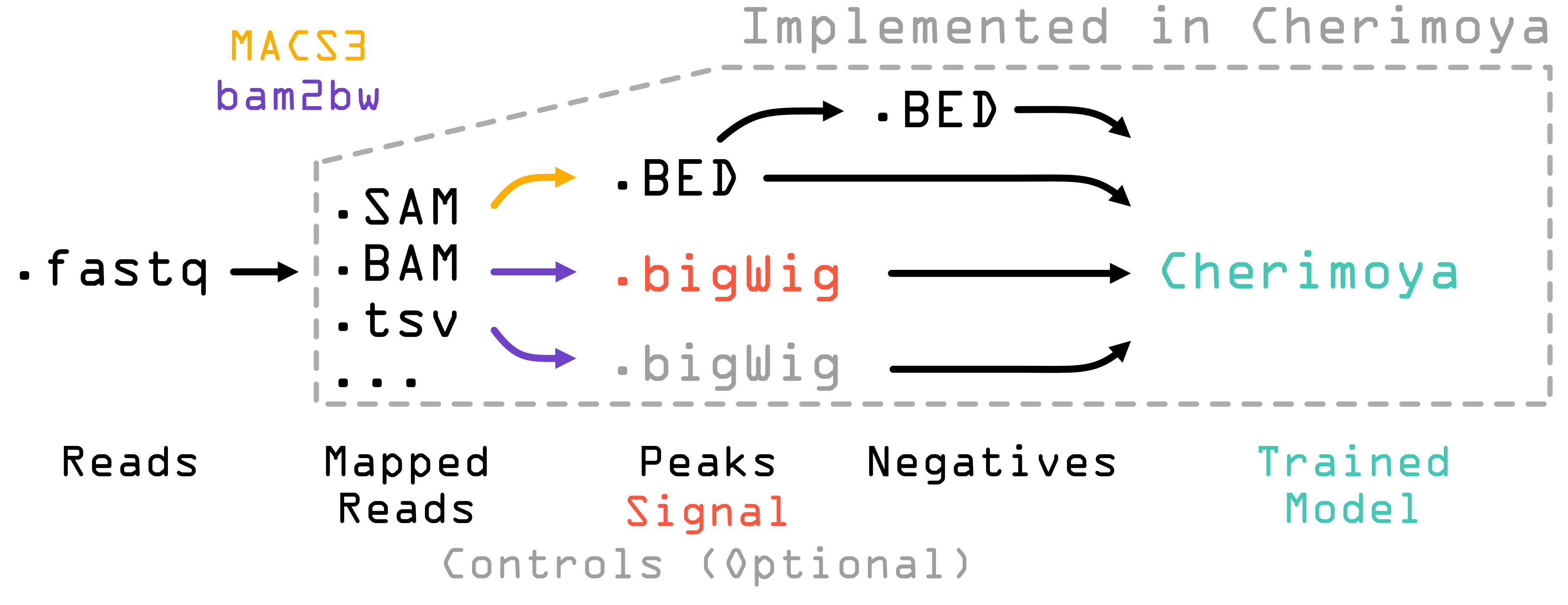
For a complete list of every CLI flag, every JSON key, and every default value, see CLI Reference. For assay-specific recipes (TF ChIP-seq, ATAC-seq, DNase-seq) see the recipe pages.
Prerequisites¶
You will need:
Cherimoya installed (see Installation).
A reference genome FASTA file (e.g.
hg38.fa).One or more signal files (BAM/SAM, fragment BED/TSV, or bigWig).
A motif database in MEME format (optional, used by TF-MoDISco reports and marginalization).
Optional: a BED of peak coordinates. If you don’t provide one, the pipeline calls peaks with MACS3.
Optional: BED of GC-matched negative regions. If you don’t provide one, the pipeline samples them automatically.
All file inputs can be remote URLs (http://, https://,
s3://, gs://). bam2bw streams the data directly without
downloading the file first; the validation step before the run only
checks paths that look local.
Subcommands at a glance¶
The two commands you actually run, in the order you run them:
cherimoya pipeline-json— emit a fully-populated JSON config from a handful of CLI pointers.cherimoya pipeline— run the full end-to-end pipeline from that JSON.
Every other subcommand (fit, evaluate, attribute,
seqlets, marginalize, negatives, batch) corresponds
to an individual pipeline stage and can be run on its own. Each is
driven by its own JSON, which the pipeline writes alongside its
outputs. See CLI Reference for the full subcommand reference.
Two-step workflow¶
The expected workflow is two steps: generate a JSON, then run it.
Step 1: generate a pipeline JSON¶
For stranded ChIP-seq with input controls:
cherimoya pipeline-json \
-s hg38.fa -p peaks.narrowPeak \
-i input1.bam -i input2.bam \
-c control1.bam -c control2.bam \
-m JASPAR_2024.meme -n my_experiment -o pipeline.json
For unstranded paired-end ATAC-seq with the standard +4 / -4 fragment shift:
cherimoya pipeline-json \
-s hg38.fa -p peaks.narrowPeak \
-i fragments.bam -m JASPAR_2024.meme \
-n atac_experiment -o pipeline.json \
-ps 4 -ns -4 -u -f -pe
Repeating -i adds another signal file. Repeating -c adds
another control. Repeating -p adds another peak file. The full
pipeline-json flag list is in CLI Reference.
The resulting JSON has every parameter the pipeline uses, filled in
from cherimoya_cli.defaults.default_pipeline_parameters. You only
need to edit the values you want to change from defaults; the runtime
re-merges with defaults before each step.
Step 2: edit and run¶
Open the JSON and override any defaults — model width, training and validation chromosomes, seqlet p-value threshold, MoDISco settings, anything in the JSON.
cherimoya pipeline -p pipeline.json
What this does, in order:
MACS3 peak calling (skipped if
lociis set).bam2bw conversion to bigWig (skipped if signals are already bigWigs).
GC-matched negative sampling (skipped if
negativesis set).Model training — writes
{name}.torch(best checkpoint by validation count Pearson) and{name}.final.torch(EMA weights at end of training), plus{name}.log.Attribution via saturation mutagenesis over the central 400 bp of each example, saved as
{name}.attributions.{ohe,attr}.npzand{name}.attributions.idxs.npy.Seqlet identification with TF-MoDISco-style recursive seqlet calling on the (attribution × one-hot) signal, written to
{name}.seqlets.bed.tomtom-lite seqlet annotation against the motif database, if
motifswas provided; results in{name}.seqlets_annotated.bedand a counts table in{name}.motif_seqlet_count.tsv.TF-MoDISco motif discovery and HTML report; results in
{name}_modisco_results.h5and{name}_modisco/.Marginalization — measures the predicted effect of inserting each motif into negative backgrounds. Output in
{name}_marginalize/.
Each sub-step writes its own JSON snapshot
({name}.fit.json, {name}.attribute.json, …) so individual
stages can be re-run in isolation, and pre-existing snapshots can be
edited and re-run if you only need to change one stage.
Running individual steps¶
Each stage has its own subcommand and JSON schema. You can run them directly:
cherimoya fit -p my_experiment.fit.json
cherimoya evaluate -p my_experiment.evaluate.json
cherimoya attribute -p my_experiment.attribute.json
cherimoya seqlets -p my_experiment.seqlets.json
cherimoya marginalize -p my_experiment.marginalize.json
The defaults for each command are in
cherimoya_cli.defaults.default_*_parameters; the merged JSON
snapshots written by pipeline make them concrete.
Every step JSON also supports "skip": true to no-op that step.
Batch mode¶
For training the same configuration across many datasets in parallel on multiple GPUs:
cherimoya batch -p batch.json
Minimal batch.json:
{
"name": null,
"device": "*",
"signals": "/path/to/data/*.bam",
"sequences": "/path/to/hg38.fa"
}
Behavior:
"device": "*"is expanded to the full list of available CUDA devices (cuda:0,cuda:1, …).When
"signals"is a glob and"name"isnull, names are derived from the signal filenames automatically.Each derived job is written to its own
{name}.pipeline.jsonand run viacherimoya pipeline. Joblib distributes jobs round-robin across the device list.
To use batch mode with custom names or paired-up controls/loci/negatives,
provide same-length lists in those fields; element i is consumed by
job i.
About bam2bw¶
The pipeline does not call BAM/SAM/fragment files directly into the
training step — it converts them to bigWig first using the
bam2bw tool, which is a hard dependency. bam2bw streams the
input (local or remote URL), counts reads or fragments into per-base
coverage, optionally applies a ± shift (used for Tn5 / DNase
corrections), and emits one bigWig (unstranded) or two bigWigs
(+ / - stranded).
This conversion is what enables remote URLs as inputs: bam2bw
fetches reads via byte-range requests rather than downloading the
whole file. The resulting bigWigs are written into the working
directory and re-used by every downstream stage.
If your signals are already bigWigs, the pipeline skips this step
automatically. To use bam2bw standalone (outside the pipeline),
invoke it directly — see its own documentation.
Calling negatives independently¶
cherimoya pipeline calls negatives for you when the JSON’s
negatives field is null. If you want to sample GC-matched
negatives without running the full pipeline (e.g. you’re going to
train a non-Cherimoya model on the same regions), use the
negatives subcommand:
cherimoya negatives \
-i peaks.narrowPeak \
-f hg38.fa \
-b signal.bw \
-o negatives.bed \
--bin_width 0.02 --max_n_perc 0.1 --beta 0.5
The output is a 3-column BED of regions matched by GC content to the
input peaks, with at most --max_n_perc fraction of N bases
and (optionally) signal below --beta × min(peak_counts).
Running on a non-hg38 reference¶
The defaults assume hg38. To run on a different reference (mouse mm10, non-human, or a different hg version), override three keys in the pipeline JSON:
fit_parameters.training_chroms— chromosomes used to train. Replace with the appropriate list for your reference (e.g. mm10:["chr1", "chr2", …, "chr19", "chrX", "chrY"]minus the two validation chromosomes you choose).fit_parameters.validation_chroms— held-out chromosomes for validation. Two chromosomes is enough.preprocessing_parameters.callpeaks_gsize— MACS3 effective genome size. Use"mm"for mouse,"ce"for C. elegans,"dm"for fly, or a numeric value (e.g."2.7e9"for hg38) for any other organism.
For non-chromosome reference contigs (scaffolds, alternate
haplotypes, viral integrations), exclude them by listing them
explicitly in the training/validation chromosome lists, or by passing
an exclusion_lists BED.
Outputs¶
A successful pipeline run leaves the following in the working
directory (with {name} from the -n flag in step 1):
File |
Contents |
|---|---|
|
Best-by-validation-count-Pearson checkpoint (config + state_dict). |
|
Final EMA-applied checkpoint at end of training. |
|
Per-epoch training and validation metrics (TSV). |
|
Final held-out chromosome metrics (single row TSV). |
|
bigWigs produced by |
|
bigWig produced by |
|
bigWigs produced by |
|
Peaks called by MACS3 (when |
|
GC-matched negative regions (when |
|
One-hot encoded sequences over the central 400 bp window. |
|
Hypothetical importance scores (saturation mutagenesis). |
|
Boolean mask into the original loci list selecting examples that had no Ns over the window. |
|
Recursive seqlets in genome coordinates. |
|
Seqlets with closest-motif annotation from tomtom-lite. |
|
Count of seqlets matched per motif. |
|
TF-MoDISco pattern HDF5. |
|
TF-MoDISco HTML report. |
|
Motif marginalization report (HTML plus CSVs). |
|
Per-step JSON snapshots of the actual parameters used. |